





Les amélogenèses imparfaites de A à Z
Clinique Par Marie-Cécile Manière, François Clauss, Elise Pilavyan, Agnès Bloch-Zupan le 11-06-2019Les amélogenèses imparfaites (AI) sont considérées comme des maladies rares à expression bucco-dentaire. Ce sont des affections d’origine génétique qui touchent toutes les dents, c’est-à-dire les dents temporaires ou dents de lait, ainsi que les dents permanentes ou définitives. Les personnes qui en sont atteintes présentent un préjudice esthétique, fonctionnel et psycho-social majeur. Ce sont des affections encore méconnues, qui représentent un véritable défi pour le chirurgien-dentiste en raison du diagnostic difficile et de la complexité de la prise en charge, qui s’organise sur de nombreuses années, de la petite enfance à l’âge adulte.
L’émail dentaire est un tissu minéralisé extrêmement résistant, composé à 98% d’une phase minérale et à 2% d’une phase organique. C’est le tissu le plus dur de l’organisme. Il est formé grâce à des cellules hautement spécialisées, les améloblastes, qui sont régulées par des voies de signalisation complexes.
Le processus d’amélogenèse est limité dans le temps et comporte différents stades (élaboration de la matrice de l’émail, minéralisation et concomitamment élimination organisée des protéines de la matrice de l’émail lors de la maturation pour aboutir à une minéralisation optimale). Il débute pendant la vie intra-utérine. L’amélogenèse est contrôlée par des gènes codant pour des protéines spécifiquement amélaires ou non; elle est influencée par des facteurs épigénétiques et environnementaux.
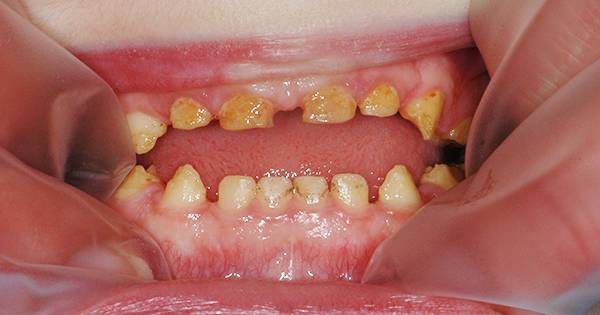
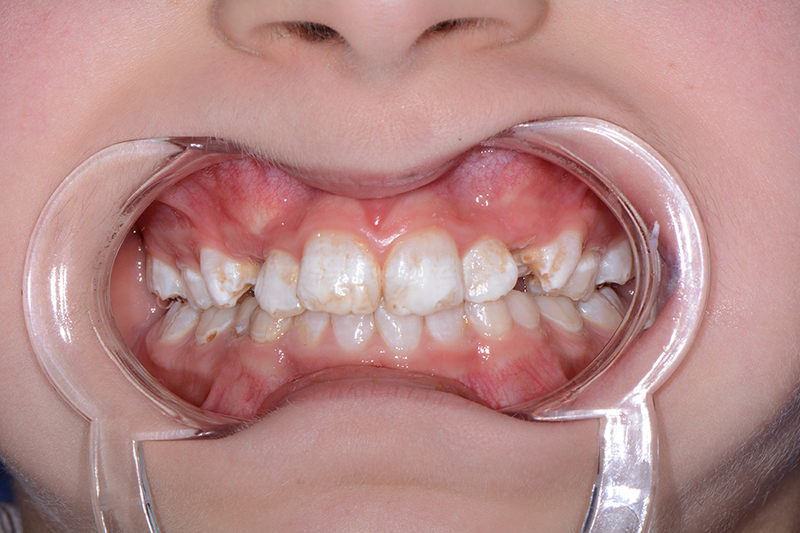
Une perturbation de l’amélogenèse peut se traduire par une réduction de la quantité d’émail formé et/ou une altération de sa minéralisation et de sa maturation, ce qui explique les différences observées en clinique.
Les aspects cliniques et radiographiques permettent de caractériser les défauts de l’émail et d’orienter le diagnostic différentiel vers une étiologie génétique ou acquise. De ce fait les antécédents personnels et familiaux doivent être systématiquement recherchés lors de la consultation.
Le nombre de dents atteintes ainsi que la localisation du défaut (symétrie…) permettent de distinguer :
1) Une anomalie isolée de l’émail telle que l’atteinte d’une seule dent traduisant généralement une étiologie locale : séquelle d’un traumatisme ou d’une infection de la dent temporaire.
2) Une atteinte d’un groupe de dents, dont la période de minéralisation est concomitante traduit une étiologie essentiellement générale : toxicité environnementale (tétracyclines, dioxine, autres polluants, radiations, bisphénol A…) ou systémique (hypoxie néonatale, infections, carences…). Dans ces cas, il est possible d’observer un large spectre de défauts amélaires, allant de l’opacité discrète, circonscrite ou diffuse, d’une seule dent, à l’hypoplasie sévère au niveau de plusieurs dents.
3) Une atteinte de toutes les dents et des deux dentures est le signe d’une étiologie génétique. Il s’agit des amélogenèses imparfaites (AI).
Les amélogenèses imparfaites représentent un groupe hétérogène d’altérations de l’émail d’origine génétique. Leur prévalence varie de 1/700 à 1/14000 selon les études et tous les modes de transmission ont été décrits, autosomique dominant, récessif, lié à l’X ; des cas sporadiques ont également été rapportés. Le processus de formation de l’émail est altéré et conduit à la modification de la structure et de l’apparence clinique de l’émail.
Selon le type de défaut principal, l’AI peut être qualifiée d’hypoplasique, d’hypominéralisée ou d’hypomature. Plusieurs formes peuvent coexister dans une même famille, chez un même patient, voire sur une même dent, ce qui complique le diagnostic. L’expression des nombreux gènes, et l’implication de différentes protéines expliquent ces formes d’AI si différentes (Tableau I).
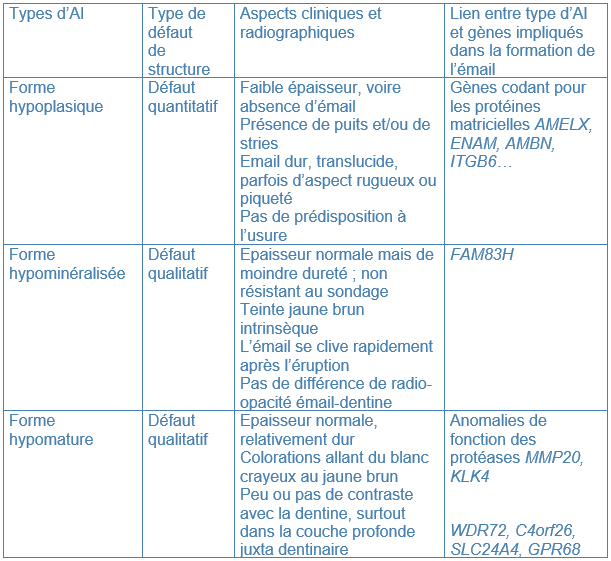
Tableau I Différentes formes d’AI et gènes impliqués
Ces anomalies peuvent exister de façon isolée. Mais elles sont parfois associées à d’autres symptômes, dans ce cas on parle d’AI syndromique. En voici quelques exemples :
• syndrome ERS (Email-Rein) : AI et néphrocalcinose
• syndrome de Jalili : AI et atteinte oculaire (dystrophie des cônes et des bâtonnets)
• syndrome de Morquio : AI et atteinte osseuse
• syndrome de Kohlschütter-Tonz : AI, épilepsie et déficience intellectuelle.
Le centre de référence des maladies rares orales et dentaires, site coordinateur de Strasbourg, a mis au point un test génétique spécifique des anomalies bucco-dentaires (panel de gènes GenoDENT, séquençage à haut débit) qui permet aux patients atteints d’AI de bénéficier d’un diagnostic moléculaire.
Les conséquences cliniques des AI sont multiples et potentiellement sévères :
• dyschromie : c’est l’anomalie de teinte des dents, par exemple des taches blanchâtres, un aspect jaunâtre, voire brunâtre des dents
• hypersensibilité (douleurs au froid, au brossage, à la mastication par exemple)
• pertes de substance.
De plus, on retrouve fréquemment :
• une malocclusion (les mâchoires ne s’emboitent pas bien, les dents sont mal alignées)
• une gingivite (inflammation des gencives)
• des troubles de l’éruption (des dents restent incluses)
• des troubles de résorption
• des anomalies dentaires de forme comme un taurodontisme (anomalie de forme des chambres pulpaires)
• plus rarement des anomalies dentaires de nombre, forme : des agénésies, une microdontie, des calcifications intra-pulpaires…
La malocclusion est fréquemment associée aux formes hypoplasiques et se manifeste principalement par une infraclusion antérieure (« béance »), qui peut être sévère, venir encore aggraver le préjudice esthétique et engendrer des difficultés de mastication et d’élocution.
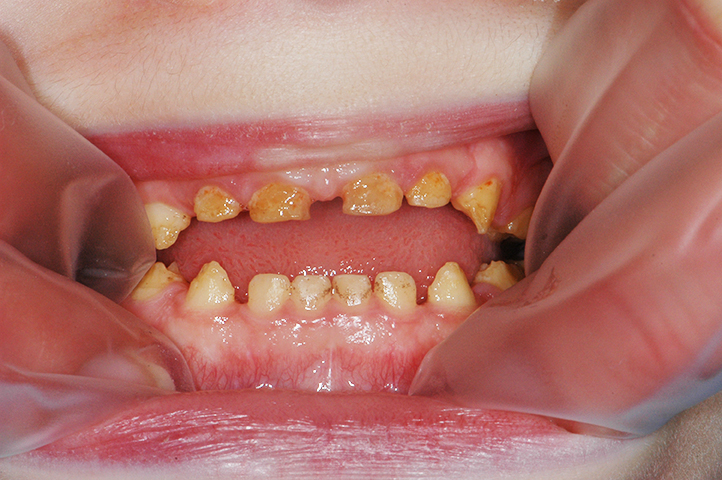
Dans la plupart des formes d’AI, l’aspect esthétique est fortement compromis. En effet, l’émail étant très fin, voire absent, la couleur de la dentine devient apparente, conférant à la dent une teinte jaunâtre voire brunâtre. Enfin, l’usure prématurée de l’émail et les pertes de substance associées à une rétention accrue de plaque sont susceptibles d’augmenter le risque carieux.
La qualité de vie des patients atteints d’AI est notablement diminuée, les patients présentent une faible estime de soi. Les enfants souffrent de moqueries dès l’école primaire, voire de harcèlement moral.
Ainsi les amélogenèses imparfaites sont des affections invalidantes car elles entrainent un préjudice esthétique et fonctionnel souvent majeur. L’impact social et économique est important du fait des traitements lourds et évolutifs, de la petite enfance à l’âge adulte.
La prise en charge thérapeutique comporte 3 volets : une approche psychologique adaptée et une prise en charge à la fois préventive et thérapeutique.
• Sur le plan psychologique, le praticien doit bien prendre en compte le contexte émotionnel du patient atteint d’une telle anomalie ; celui-ci vient consulter alors qu’il a déjà souffert de douleurs et a pris conscience dès son plus jeune âge de l’aspect inesthétique de ses dents.
Le recours à la sédation consciente ou à l’anesthésie générale se justifie dans la mesure où les patients traités sont jeunes, les séances de soins nombreuses et longues, en raison du nombre élevé de dents atteintes et de la sévérité des lésions. Par ailleurs, les hypersensibilités rendent l’anesthésie locale de la dent souvent difficile. De plus, dans les AI syndromiques et dans le cas de troubles de l’éruption, l’intervention d’un chirurgien maxillo-facial est souvent indiquée.
• Prise en charge préventive
L’enseignement de l’hygiène bucco-dentaire est capital : l’émail anormal favorise la rétention de plaque, un brossage correct est donc indispensable pour limiter l’inflammation gingivale. Le brossage s’avère souvent douloureux et il est nécessaire de restaurer les dents avant d’envisager un bon contrôle de plaque.
Le traitement préventif a aussi pour objectif d’améliorer la minéralisation de l’émail. La fluoration, grâce à des applications topiques en cabinet et une utilisation régulière de dentifrice fluoré au domicile, peut contribuer à diminuer l’hypersensibilité. D’autres techniques comme l’érosion/infiltration résineuse au niveau des faces lisses ou les scellements de sillons occlusaux sont également indiquées.
• Sur le plan thérapeutique, les solutions esthétiques et fonctionnelles doivent être durables et prédictibles.
Quel que soit l’âge, une prise en charge multidisciplinaire est indispensable et fait intervenir des spécialités comme l’odontologie pédiatrique, l’orthopédie dento-faciale, l’endodontie, la parodontologie, la prothèse parfois la chirurgie maxillo-faciale, notamment pour les cas complexes comportant une béance antérieure importante.
Les traitements orthodontiques sont un des volets de la thérapeutique. Les retards d’éruption, les rétentions dentaires et les anomalies radiculaires viennent compliquer cette prise en charge orthodontique.
En denture temporaire et durant la croissance, les traitements restaurateurs et prothétiques sont transitoires et évolutifs. Ils dépendent notamment du degré d’atteinte tissulaire. Les défauts plus sévères seront traités par des coiffes pédodontiques, soit métalliques préformées pour les dents postérieures, soit en composite de laboratoire, soit en Zircone. Un suivi régulier tous les 6 mois est impératif afin de garantir la pérennité des restaurations.
Chez l’adolescent et l’adulte, de même les thérapeutiques sont fonction de la gravité de l’AI et s’étalent sur de nombreuses années:
• L’anesthésie locale est indispensable, quel que soit le traitement.
• La micro-abrasion peut être utilisée pour éliminer les défauts superficiels de l’émail tels que les opacités blanchâtres ou brunes.
• Les techniques d’éclaircissement en cabinet (« blanchiment ») visent également à réduire certaines dyschromies, mais ne peuvent pas s’appliquer à tous les cas et sont réservées aux patients âgés de plus de 18 ans.
• Dans les cas d’anomalies plus sévères, des restaurations adhésives à l’aide de composites ou de ciments verre-ionomère peuvent être indiquées pour restaurer transitoirement des molaires présentant une hypersensibilité.
• En denture permanente, les inlay-onlay, facettes et couronnes céramiques constituent les techniques de choix.
La longévité des restaurations au composite est moindre chez les patients souffrant d’AI. De récentes études ont montré l’efficacité, tant sur le plan fonctionnel que de la qualité de vie, d’un traitement prothétique par couronnes céramiques chez l’enfant et l’adolescent présentant une forme sévère d’AI. Enfin, grâce au développement actuel des techniques de CFAO (« conception assistée par ordinateur »), les patients atteints d’AI peuvent bénéficier des derniers progrès en matière de réhabilitation prothétique.
Conclusion
Les amélogenèses imparfaites sont des maladies rares d’origine génétique potentiellement invalidantes. Elles entrainent des préjudices esthétique, fonctionnel et psycho-social sévères.
Elles doivent être prises en charge le plus précocement possible afin de préserver le capital dentaire et de rétablir l’esthétique et des fonctions orales correctes. L’épanouissement psycho-social de l’enfant et son bien-être en dépendent !
Compte tenu de la complexité de la prise en charge, le patient doit souvent être dirigé vers une ressource hospitalière spécialisée, comme un centre de compétence ou un centre de référence maladies rare, Réseau O-Rares, ce qui peut engendrer des coûts supplémentaires. La collaboration avec le chirurgien-dentiste traitant est essentielle. Le centre maladie rare peut notamment intervenir pour une assistance au diagnostic et/ou un avis thérapeutique. Il est très important de préciser le caractère isolé ou syndromique de l’AI. Un recours au bilan, diagnostic et conseil génétique en collaboration avec le médecin généticien est utile.
Le reste à charge pour le patient, en raison de ces traitements complexes de longue haleine, reste élevé. Il n’existe pas encore d’ALD hors liste, comme celle qui a été mise en place pour l’oligodontie, autre maladie génétique bucco-dentaire, dans le cadre du premier plan national maladies rares.
Bibliographie
Crawford PJ, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta.
Orphanet J Rare Dis. 2007; 4:2:17
de la Dure-Molla M, Quentric M, Yamaguti PM, Acevedo AC, Mighell AJ, Vikkula M, Huckert M, Berdal A, Bloch-Zupan A. Pathognomonic oral profile of Enamel Renal Syndrome (ERS) caused by recessive FAM20A mutations.
Orphanet J Rare Dis. 2014; 9:84.
C Naulin Ifi. Odontologie pédiatrique clinique. Ed CdP, Paris, 2011
Huckert M, Mecili H, Laugel-Haushalter V, Stoetzel C, Muller J, Flori E, Laugel V, Manière MC, Dollfus H, Bloch-Zupan A. A Novel Mutation in the ROGDI Gene in a Patient with Kohlschütter-Tönz Syndrome.
Mol Syndromol. 2014; 5(6):293-8.
Huckert M, Stoetzel C, Morkmued S, Laugel-Haushalter V, Geoffroy V, Muller J, Clauss F, Prasad MK, Obry F, Raymond JL, Switala M, Alembik Y, Soskin S, Mathieu E, Hemmerlé J, Weickert JL, Dabovic BB, Rifkin DB, Dheedene A, Boudin E, Caluseriu O, Cholette MC, Mcleod R, Antequera R, Gellé MP, Coeuriot JL, Jacquelin LF, Bailleul-Forestier I, Manière MC, Van Hul W, Bertola D, Dollé P, Verloes A, Mortier G, Dollfus H, Bloch-Zupan A. Mutations in the latent TGF-beta binding protein 3 (LTBP3) gene cause brachyolmia with amelogenesis imperfecta.
Hum Mol Genet. 2015; 24(11):3038-49.
Lundgren GP, Vestlund GM, Dahllöf G. Crown therapy in young individuals with amelogenesis imperfecta: Long term follow-up of a randomized controlled trial.
J Dent. 2018; 76:102-108
Parekh S, Almehateb M, Cunningham SJ. How do children with amelogenesis imperfect feel about their teeth?
Int J Paed Dent. 2014; 24(5): 326-335
Pousette Lundgren G, Hasselblad T, Johansson AS, Johansson A, Dahllöf G. Experiences of Being a Parent to a Child with Amelogenesis Imperfecta.
Dent J (Basel). 2019; 7(1)
Rey T, Tarabeux J, Gerard B, Delbarre M, Le Béchec A, Stoetzel C, Prasad M, Laugel-Haushalter V, Kawczynski M, Muller J, Chelly J, Dollfus H, Manière MC, Bloch-Zupan A.
Protocol GenoDENT: Implementation of a New NGS Panel for Molecular Diagnosis of Genetic Disorders with Orodental Involvement.
Methods Mol Biol. 2019; 1922:407-452.
Strauch S, Hahnel S. Restorative Treatment in Patients with Amelogenesis Imperfecta: A Review.
J Prosthodont. 2018; 27(7):618-623
Toupenay S, Fournier BP, Manière MC, Ifi-Naulin C, Berdal A, de La Dure-Molla M. Amelogenesis imperfecta: therapeutic strategy from primary to permanent dentition across case reports.
BMC Oral Health. 2018; 18(1):108
Pour aller plus loin, retrouvez le replay du webinar Colgate sur le thème :
Les amélogenèses imparfaites de A à Z